
北京健平金星生物医药

新闻资讯
自噬是细胞在外界环境因素的影响下,利用溶酶体,降解自身受损、变性大分子物质或者细胞器的自我消化过程。依据其发生途径,主要分为三种:巨自噬 (Macroautophagy),微自噬 (Microautophagy) 和分子伴侣介导的自噬 (Chaperone-mediated autophagy, CMA)。
巨自噬过程中,自噬体的双膜囊泡将受损的蛋白质/细胞器传递至溶酶体进行降解,通常文献中提到的自噬指的是巨自噬。微自噬也通过囊泡将物质输送到溶酶体,溶酶体膜内陷,降解方式直接了当。
分子伴侣介导的自噬:与前两种自噬不同,伴侣介导的自噬不使用囊泡,这种自噬方式具有高度选择性,常借助伴侣蛋白 Hsc70,特异性降解带有独特识别五肽基序 (KFERQ 样) 的靶蛋白,溶酶体膜上的受体蛋白 LAMP2A 识别结合蛋白暴露的 KFERQ 基团,“引导”目的蛋白进入溶酶体降解。
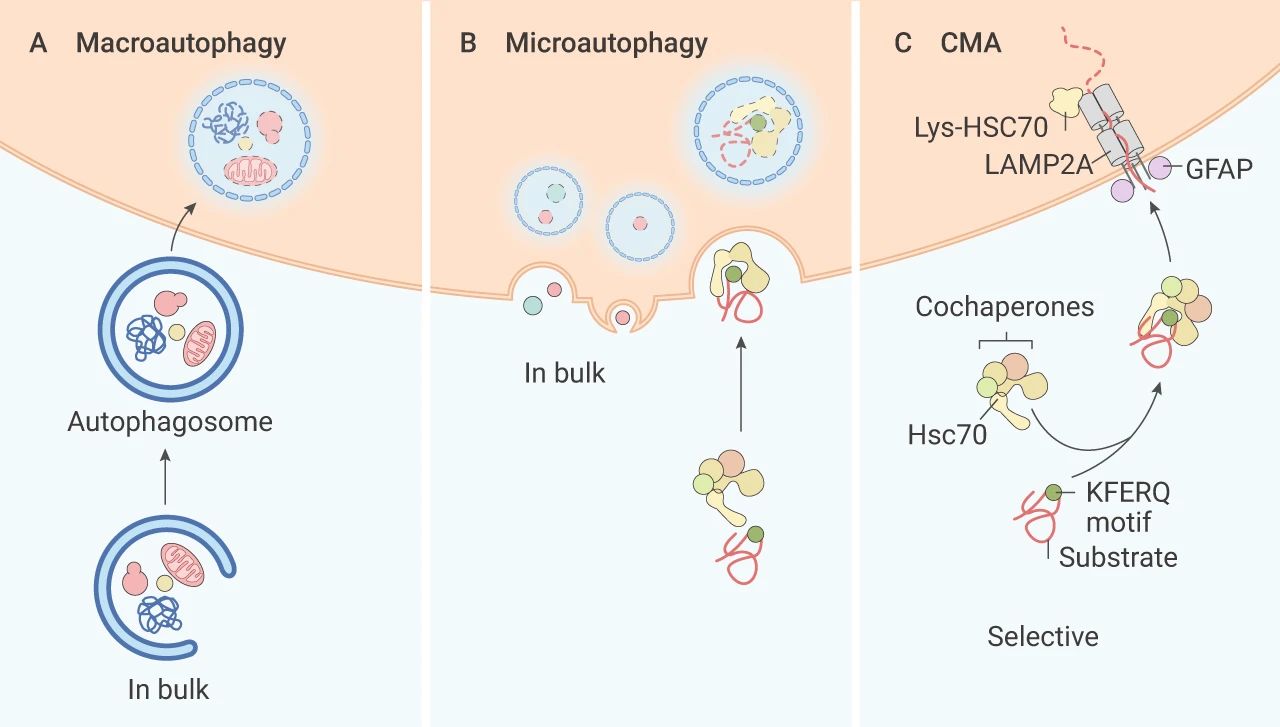
图 1. 三种不同的自噬途径[2]
A. 巨自噬;B. 微自噬;C. 伴侣介导的自噬
神经退行性疾病中,神经细胞往往会累积大量无法降解的蛋白,通过溶酶体自噬过程降解蛋白是细胞清除异常蛋白的主要方式。已有报道,CMA 涉及神经退行性疾病病原蛋白质 (如 α-syn 和 tau) 的降解,然而关于神经退行性疾病中 CMA 的失去功能导致的后果尚未清楚。近日,Ana Maria Cuervo 等人在Cell上发表了题为Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome的文章,揭示了 CMA 对神经细胞稳态的重要调节作用。

在这篇文章中,研究者们分别构建了全身性 LAMP2A (L2A) 敲除模型 (L2A-/-) 和神经元条件敲除 L2A/LAMP2a (CKL2A-/-) 小鼠模型 (图 3A),来分析 CMA 对维持神经元蛋白质稳态的作用。还建立了模拟巨自噬缺失(ATG7 的条件性敲除)CKATG7-/-小鼠模型 (图 3B),并与 CKL2A-/-小鼠模型对比,来分析 CMA 和巨自噬这两条自噬通路对神经元蛋白变性的特定作用。(CTR 组小鼠为不做敲除的对照组)。
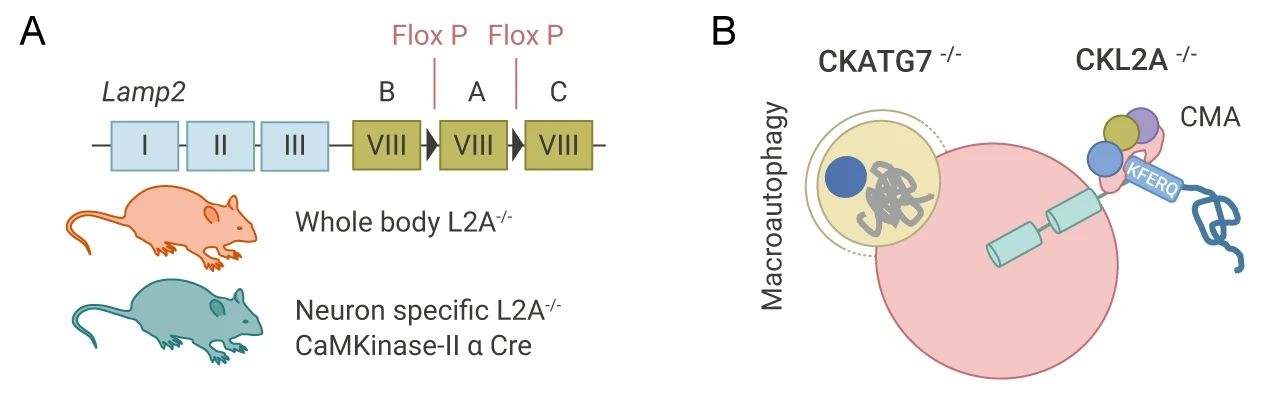
图 3.建立小鼠实验模型
A. L2A 的全身性敲除和条件敲除;B. ATG7 的条件性敲除
■CKL2A-/-小鼠和 L2A-/-小鼠所表现出的神经损害行为
首先,研究者们发现,CKL2A-/-小鼠和 L2A-/-小鼠与对照组(CTR)相比,行为学测试得分更高,后肢紧握进展更快 (图 4A),这两种小鼠还均表现出了感觉运动功能障碍和 Y 型迷宫中短时记忆减少 (图 4B-C) 。只有 L2A-/-小鼠表现出类似帕金森疾病的步态特征,如步幅减少 (图 4D)。此外,CKL2A-/-小鼠还表现出了神经变性的常见表型,如有空间工作记忆减少和筑巢行为显着减少的趋势 (图 4E-F)。以上结果显示,神经元 L2A 缺失的小鼠表现出 L2A 系统性缺失小鼠的大部分行为输出。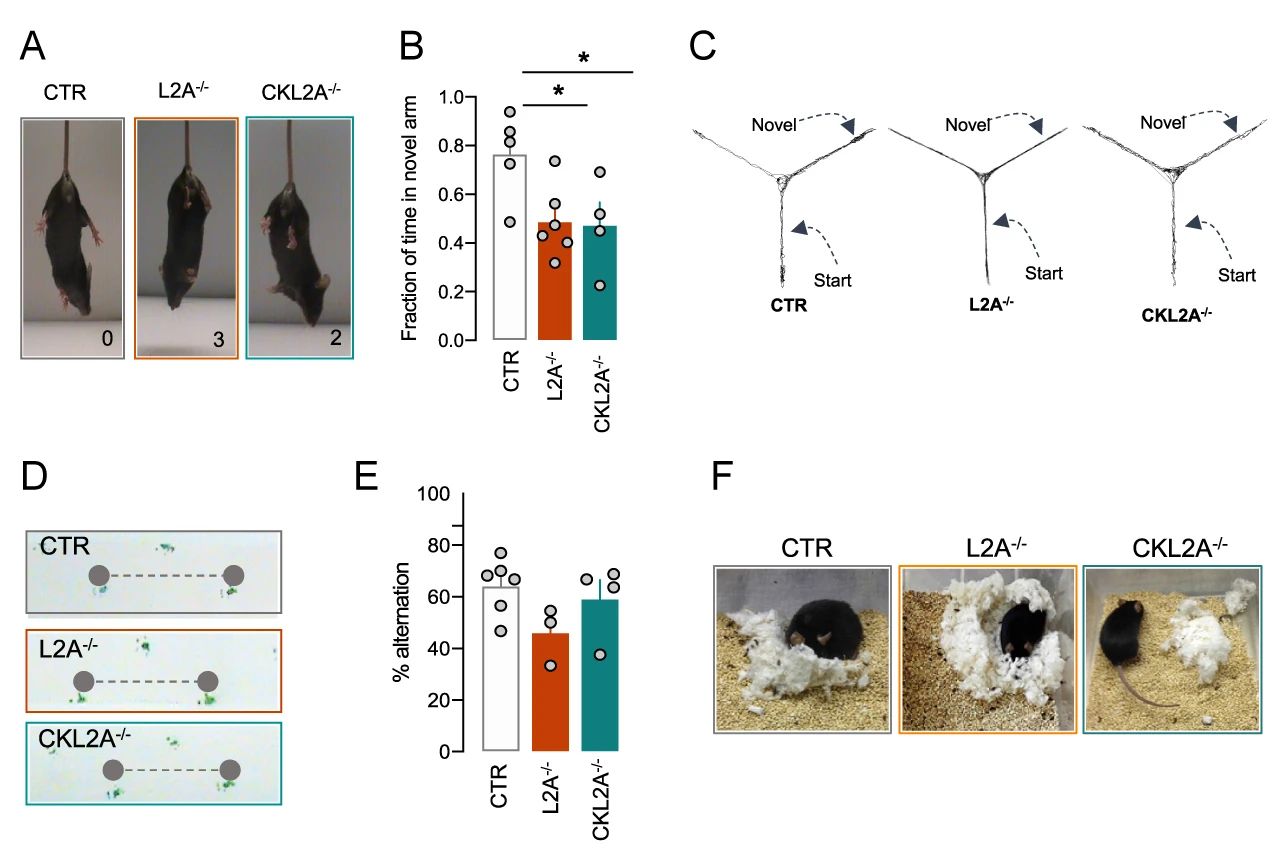
图 4.L2A 缺失小鼠表现出行为障碍A. 鼠爪抓力测定;B-C、E. Y 型迷宫行为学检测;D. 步态分析;F. 筑巢试验
■神经元 CMA 缺陷导致蛋白质稳态的破坏
作者团队发现,在 6 个月大的 L2A-/-小鼠的海马体中,出现了脂褐质和 K63 泛素化蛋白的积累 (两个蛋白是溶酶体降解的靶蛋白),CKL2A-/-小鼠的海马体的锥体神经元中也发现了类似特征 (图 5A-B)。同时 CKL2A-/-小鼠皮质的免疫印迹分析显示,氧化蛋白泛素化蛋白累积 (图 5C-D),这些结果表明CMA 缺失会导致神经元蛋白稳态的破坏。
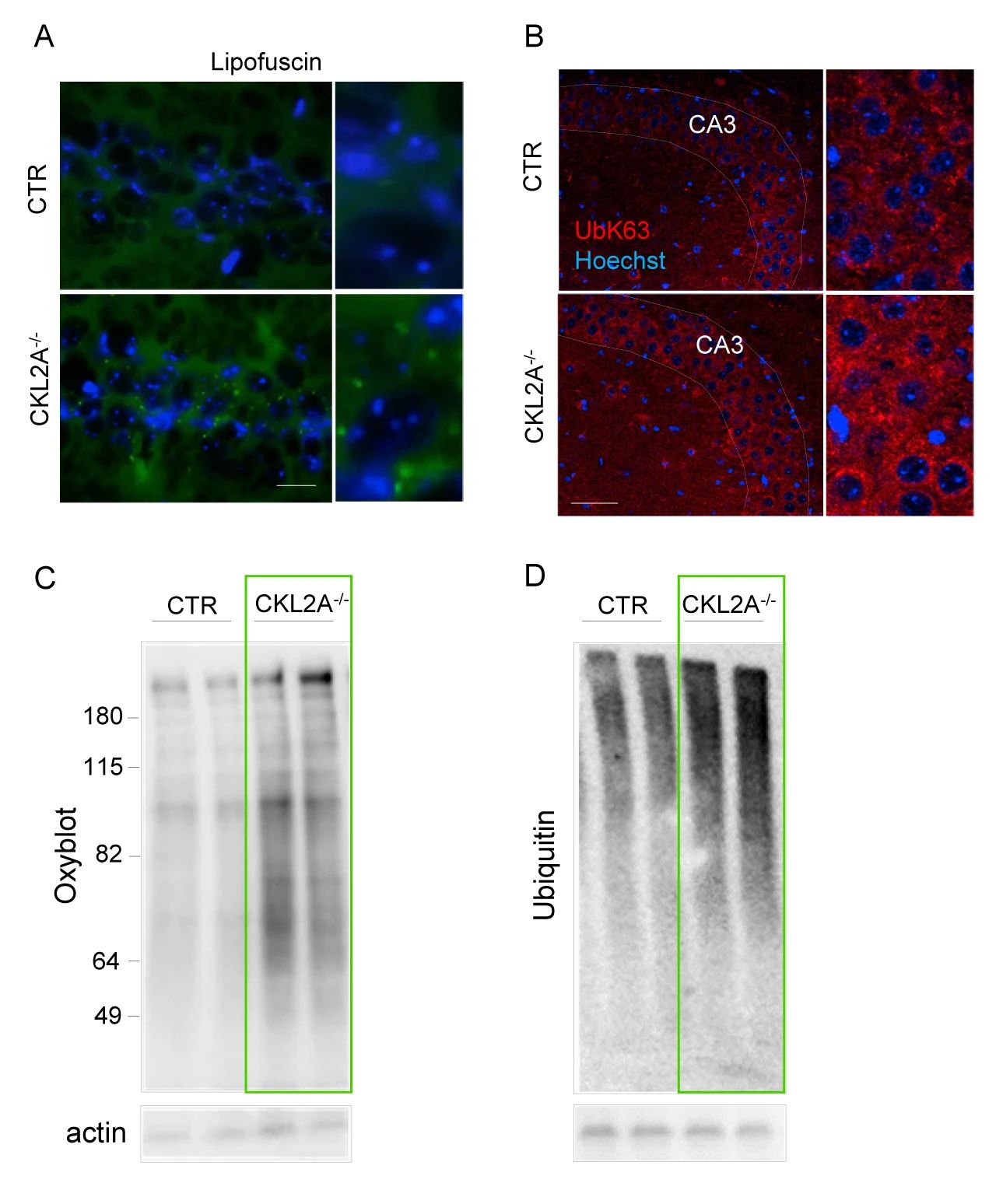
图 5.CMA 缺失会导致神经元蛋白稳态的破坏A. 海马体中的脂褐素荧光检测;B. K63 泛素化蛋白的检测;C-D. 氧化蛋白和泛素化蛋白积累的检测
紧接着,研究者们分离 CTR 组和 CKL2A-/-小鼠皮层 Sarkosy 不可溶蛋白,进行了定量蛋白组学的分析。实验结果表明 CKL2A-/-小鼠大脑聚集了大量不可溶蛋白,且这些蛋白 76% 含有 KFERQ 样基序,原本易于聚集的带有 KFERQ 样基序的蛋白质 (例如 α-syn、tau、UCHL1 和 PARK7 等蛋白) 向不溶性蛋白的转变增多 (图 6B)。研究者们还分析了过饱和分数,发现 CKL2A-/-小鼠的不溶性蛋白质中的过饱和分数显着增加 (20.37 倍)。

以上结果表明,在 CMA 缺陷时转变为不溶性的蛋白质组是过饱和蛋白质组中的一部分,并且神经元 CMA 缺失会导致蛋白质组的破坏。
■CMA 和巨自噬对神经元蛋白质变性的影响不同
下一步,研究者们在CKATG7-/-小鼠和 CKL2A-/-小鼠模型中,通过比较蛋白质组学来阐明 CMA 和巨自噬对神经元蛋白质变性的特定作用。不溶性蛋白质组的基因集富集和富集图分析 (图 7A) 结果表明,CKATG7-/-小鼠和 CKL2A-/-小鼠中发生变化的蛋白质组不相同。CKATG7-/-导致细胞周期和泛素化蛋白酶体分解代谢过程相关的蛋白变化 (蓝色箭头指出),而与 CMA 缺陷与蛋白质运输和代谢有关 (红色箭头指出)。细胞外酸化率 (ECAR) 的测定结果表明,CKL2A-/-神经元中糖酵解的显着减少 (图 7B),而且很多糖酵解酶也在 CKL2A-/-小鼠的不溶性蛋白部分中增加,如丙酮酸脱氢酶 (PDH) (图 7C)。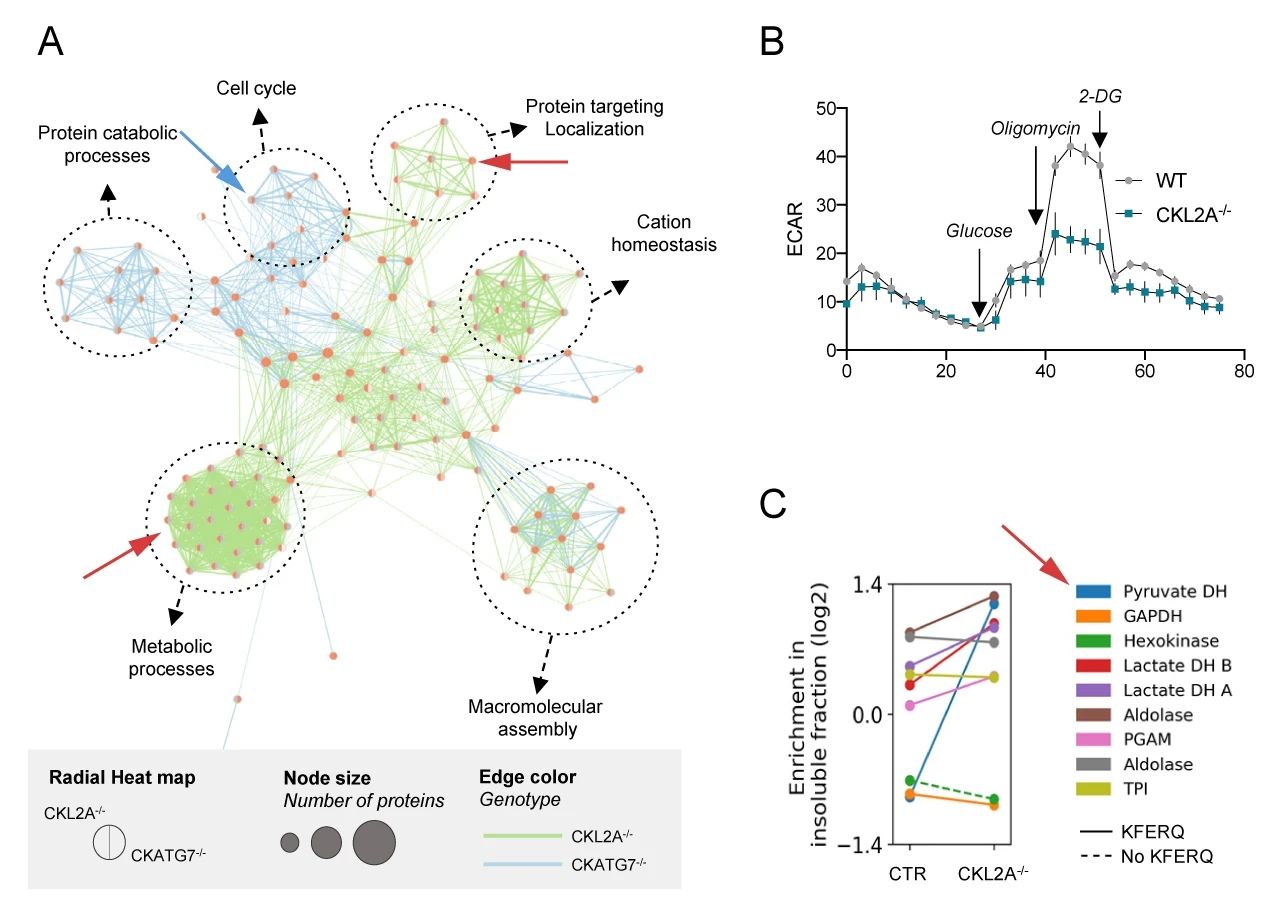
图 7.神经元蛋白质组不同亚群的变化分析
A. 不溶性蛋白基因集富集分析;B. 细胞外酸化率(ECAR)的测定;C:糖酵解酶表达的减少
虽然已有报道阻断巨自噬也会影响神经元糖酵解,但实验结果显示,L2A 和 ATG7 敲低对糖酵解特性的影响并不同。
以上结果表明,CMA 缺陷神经元蛋白质组向不溶性蛋白转变后导致的结果区别于巨自噬。基于上面的研究,推断:神经元 CMA 被直接阻断后,不溶性蛋白的积累和神经元功能的改变,这可能增加神经退行性疾病的易损性并加速疾病进展。研究者建立了 hTauP301L过表达小鼠,结果表明:其神经细胞的 CMA 活性降低,CMA 斑点数目明显减少 (图 8A)。
研究者还建立了 P301S tau 转基因小鼠,在该模型中使用了CA77.1(AR7 的衍生物) 在体外激活 CMA 且不影响巨自噬 (图 8B)。在体内,CA77.1 给药使得原本“凌乱”的 PS19 小鼠的正常化 (图 8C),并显著减少了海马、杏仁核和梨状皮层中含有致病性 tau 构象的神经元的数量 (图 8D)。
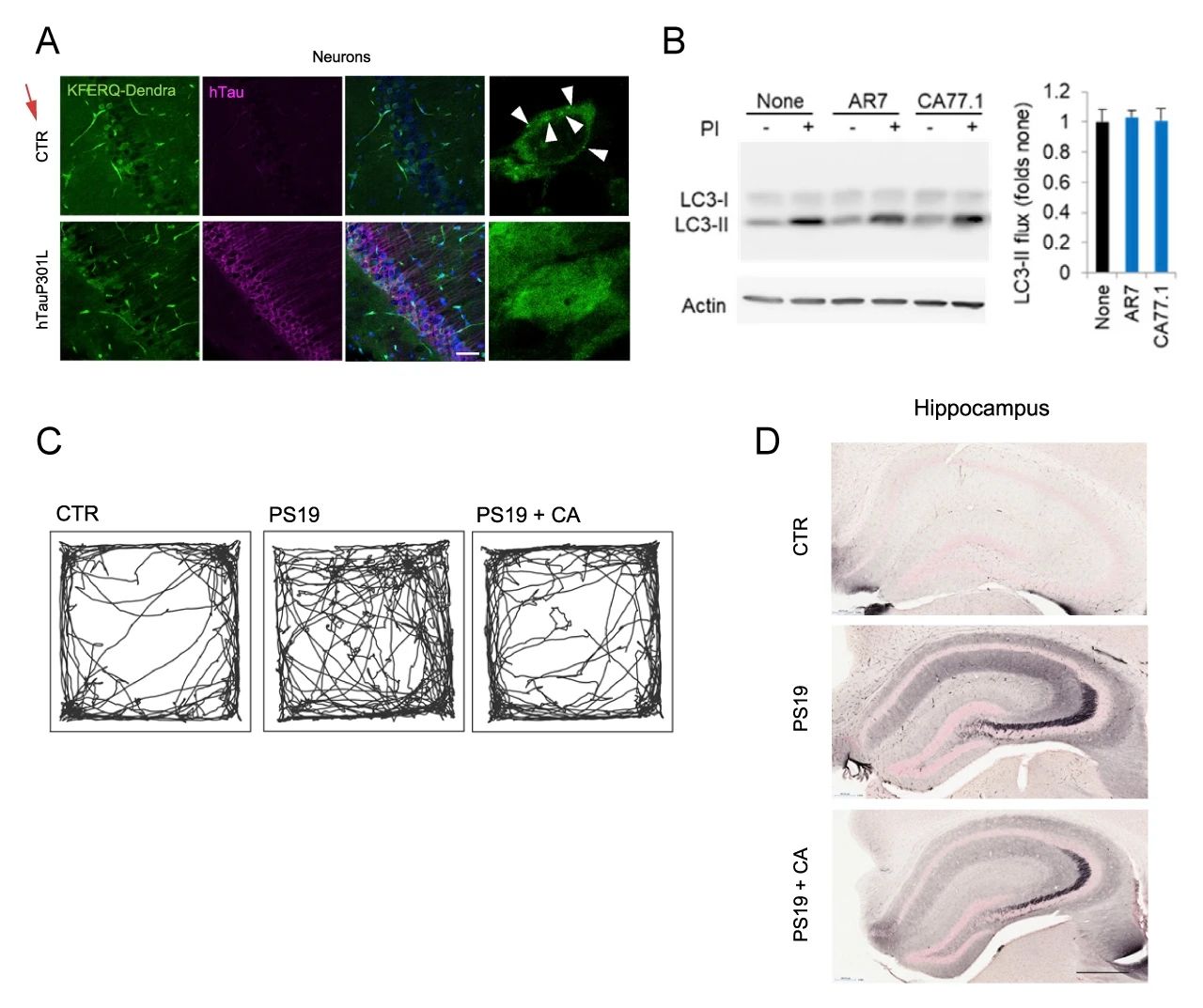
图 8.CMA 的化学激活改善了 hTauP301L 和 PS19 小鼠的神经病理特征
A. hTauP301L 小鼠神经元 CMA 染色;B. CA77.1 体外激活 CMA;C. P301S tau 小鼠的行为测试;D. 海马体免疫组化染色
总结:这篇文章充分证明了分子伴侣自噬 (CMA) 对神经细胞稳态具有重要调节作用。
研究者们通过具有全身性和神经元特异性 CMA 阻断的小鼠模型,证明了神经元 CMA 的缺失导致蛋白质毒性和神经元功能障碍。CMA 缺失还会使原本易于聚集的 KFERQ 样基序蛋白质向不溶性蛋白转变。
通过神经元特异性 CMA 和 ATG7 缺失小鼠模型,证明了 CMA 和巨自噬在调控神经元蛋白质稳态中与神经变性的亚蛋白质组并不相同。

